

Overview
The Modelling and Atomistic Simulations Laboratory within ESISNA focuses on the theoretical understanding, prediction and design of advanced functional materials through state-of-the-art atomistic simulations and first-principles calculations. Our research combines physics, chemistry, materials science and computational modelling to uncover the fundamental mechanisms governing the structural, electronic, optical, catalytic and sensing properties of nanostructured materials across multiple length scales. By integrating Density Functional Theory (DFT), molecular dynamics, machine learning and multiscale modelling approaches, we provide predictive insights that guide experimental developments in nanotechnology, surface science, energy materials, catalysis, molecular electronics, sensing and astrochemistry.
Research Goals
The main objective of our research is to establish robust theoretical frameworks capable of predicting and rationalizing the behavior of low-dimensional functional materials and complex interfacial processes.
Particular emphasis is placed on:
- Understanding structure-property relationships at the atomic scale.
- Designing novel materials with targeted functionalities.
- Predicting chemical reactivity and catalytic activity.
- Developing physically meaningful descriptors for material screening.
- Supporting experimental efforts through predictive simulations and mechanistic understanding.
- Integrating artificial intelligence with atomistic modelling for accelerated materials discovery.
Main Research Areas
Low-Dimensional Functional Materials
We investigate a broad range of nanostructured systems including:
- Graphene and graphitic nanomaterials.
- Transition-metal dichalcogenides (TMDCs).
- Two-dimensional organic materials.
- Metal-organic and covalent organic frameworks (MOFs and COFs).
- Metal-organic coordination networks.
- Nanowires, nanotubes and nanoparticles.
- Organometallic and SiC-based clusters.
- Polycyclic aromatic hydrocarbons (PAHs).
These materials are studied with the aim of understanding their structural stability, electronic properties, optical response, transport behavior and chemical functionality.

Small 2024, 20, 2312235 | Front cover Spanish Journal of Physics 2024, 38(2)
Surface and Interface Science
We explore atomistic processes occurring at surfaces and interfaces, including:
- On-surface molecular synthesis.
- Self-assembly and supramolecular organization.
- Molecule–surface interactions.
- Organic/metal and organic/oxide interfaces.
- Charge transfer phenomena.
- Surface-confined chemical reactions.
- Adsorption and desorption processes.
Special attention is devoted to the rational design of atomically precise low-dimensional architectures with tailored functionalities.

JACS 2019, 141, 3550
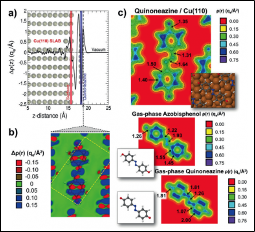
Angew. Chem. Int. Ed. 2018, 57(28), 8582-8586
Energy, Catalysis and Sustainability
Our simulations contribute to the development of advanced materials for:
- Hydrogen storage.
- Electrocatalysis and photocatalysis.
- Fuel cells and solar energy conversion.
- Environmental remediation.
- Carbon capture and utilization.
- Sustainable energy technologies.
Through first-principles modelling we identify reaction mechanisms, active sites and descriptors governing catalytic performance.

Mater. Horiz. 2025, 12, 8472-8480
Sensors and Functional Nanodevices
We develop theoretical strategies for the design of next-generation sensing platforms based on:
- Graphene and 2D materials.
- Functionalized surfaces.
- Organic frameworks.
- Hybrid nanomaterials.
- Electronic and electrochemical transducers.
Current applications include chemical sensing, environmental monitoring, biosensing and pathogen detection.

J. Mater. Chem. A 2022, 10, 4634-4643
Astrochemistry and Molecular Formation in Space
A dedicated research line focuses on understanding the atomistic mechanisms responsible for the formation and evolution of complex carbonaceous species in astrophysical environments.
Topics include:
- Polycyclic aromatic hydrocarbons (PAHs).
- Carbon-rich clusters.
- Silicon carbide clusters.
- Interstellar dust precursors.
- Surface-assisted molecular growth mechanisms.
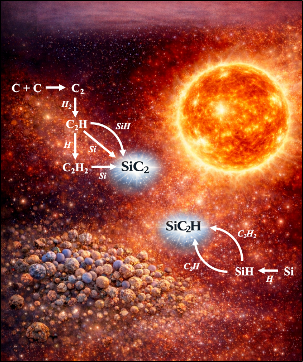
Nat. Astron. 2026, DOI: 10.1038/s41550-026-02854-1
Methodological Approach
Our research relies on advanced computational techniques including:
- Density Functional Theory (DFT).
- DFT+U and hybrid functionals.
- Molecular Dynamics (MD).
- Enhanced sampling techniques.
- Transition-state calculations (NEB and CI-NEB).
- QM/MM approaches.
- Machine Learning and Artificial Intelligence.
- High-throughput computational screening.
These methodologies allow us to investigate systems ranging from isolated molecules and clusters to extended surfaces and bulk materials.
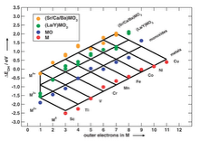
Chem. Sci. 2013, 4, 1245-1249
Predictive Materials Design
A central aspect of our work is the development of simple yet powerful descriptors that establish quantitative relationships between:
- Geometry and stability.
- Geometry and reactivity.
- Electronic structure and functionality.
- Thermodynamics and kinetics.
- Structure and catalytic activity.
These descriptors enable the accelerated identification of promising materials for targeted technological applications.
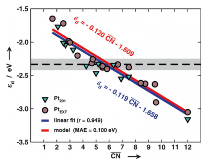
Angew. Chem. Int. Ed. 2014, 53(32), 8316-8319
Representative Research Highlights
- Atomically precise on-surface synthesis of two-dimensional organic and metal-organic networks.
- Functionalization of graphene for sensing applications.
- Hydrogen-assisted engineering of transition-metal dichalcogenides.
- Design of responsive two-dimensional coordination polymers.
- Atomistic modelling of catalytic and photocatalytic processes.
- Theoretical studies of carbonaceous species relevant to astrochemistry.
- Development of AI-assisted materials discovery workflows.
Responsible Scientist
José Ignacio Martínez Ruiz
Modelling and Atomistic Simulations Laboratory (MASLab)
Institute of Materials Science of Madrid (ICMM-CSIC)
Selected Recent Publications
- A. Rochefort*, et al., …, J. I. Martínez, Role of the Structure and Reactivity of Cu and Ag Surfaces in the Formation of a 2D Metal-Hexahydroxytriphenylene Network, Journal of Physical Chemistry C 125 (2021), 17333-17344.
- S.Gámez-Valenzuela, J. A. Alonso, G. Santoro and J. I. Martínez*, Structure, Stability, and Optical Absorption Spectra of Small TinCx Clusters: A First-Principles Approach, Monthly Notices of the Royal Astronomical Society 508 (2021), 5074-5091.
- J. I. Martínez* and J. A. Alonso, Concentration Asymmetry and Carbon Enrichment in Titanium Carbide and Silicon Carbide Clusters, Physical Review A 105 (2022), 062820.
- J. I. Martínez*, A. Laikhtman, A. Zak, M. Sezen and J. A. Alonso, Implantation of Ga into Layered WS₂ Nanostructures Is Facilitated by Hydrogenation, Small 20 (2024), 2312235.
- K. Mathialagan, et al., …, J. I. Martínez*, J. M. Gallego, N. Martín and D. Écija*, Tailoring the Magnetic Properties of 2D Metal–Organic Networks by Harnessing the Coordination Sphere, Angewandte Chemie International Edition 64 (2025), e202509199.